
da “Pillole di Ottimismo” del 27 giugno 2020 – LINK
Una domanda a cui tutti vorremmo dare una risposta è: quale sarà il futuro di SARS-CoV-2 e come potrà evolversi, soprattutto in relazione alla sua capacità di esprimersi sotto forma di una patologia più o meno letale?
Partiamo da cosa è accaduto a SARS-CoV, il virus che causò l’epidemia di SARS nel 2003 [1], tenendo presente che i due virus, SARS-CoV e SARS-CoV-2, possono essere ritenuti stretti parenti. SARS-CoV era uno dei tanti coronavirus che albergavano nei pipistrelli e che, a seguito di un processo spontaneo di mutazione, diventò in grado di infettare l’uomo [2] esattamente come è successo per SARS-CoV-2 nell’autunno-inverno 2019-2020 [3,4].
La storia della SARS che tutti ricordiamo è che la circolazione del virus si limitò ad espandersi in alcune aree geografiche con una letalità di molto superiore a quella dell’attuale SARS-CoV-2 [5, 6]. Ovviamente, molti studi sono stati portati avanti al fine di comprendere come il virus della SARS fosse cambiato geneticamente nel corso dell’epidemia.
Nel genoma dei coronavirus sono presenti geni codificanti proteine strutturali (cioè necessarie a formare la struttura del virus), proteine non strutturali, le quali hanno funzioni enzimatiche necessarie per la replicazione del virus e proteine accessorie. Nel genoma di SARS-CoV (Fig. A) sono presenti 2 grandi geni, ORF1a e ORF1b, che codificano per 16 proteine non strutturali le quali sono altamente conservate nei coronavirus (quindi anche in SARS-CoV-2); quattro sono i geni che codificano, invece, per le principali proteine strutturali (S codificante la proteina spike; E codificante envelope; M codificante membrane ed N codificante nucleocapside); gli altri geni (ORF 3, 4a, 4b, 6, 7, 8, 9) codificano per proteine accessorie che possono variare di numero, sequenza ed organizzazione genomica nei diversi coronavirus [7].
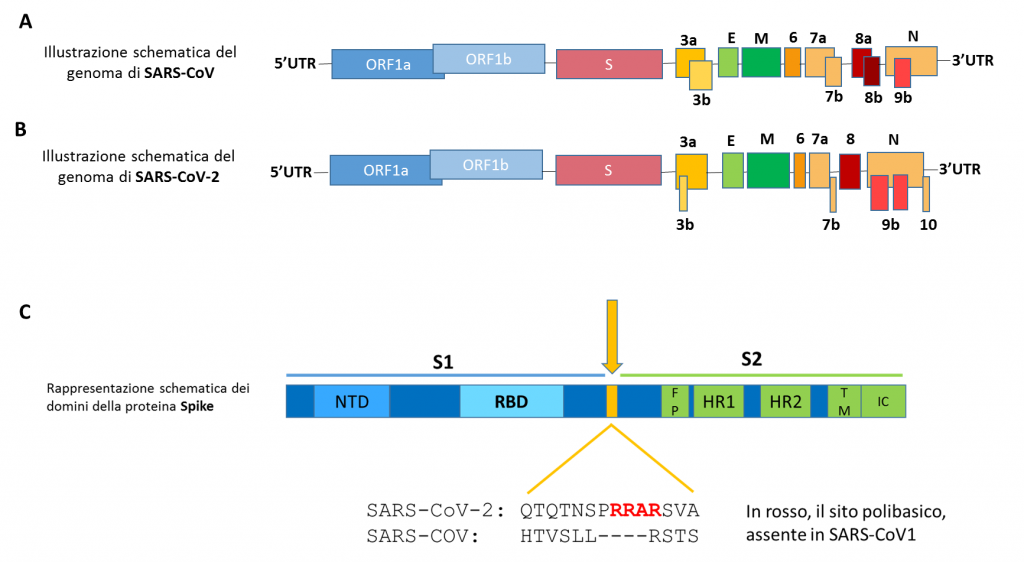
Studi di evoluzione molecolare di SARS-CoV hanno messo in evidenza come la ORF8 avesse subito sostanziali cambiamenti durante la pandemia di SARS. Nella prima fase della epidemia, SARS-CoV presentava una ORF 8 intera così come è presente in quasi tutti i coronavirus associati al pipistrello e ai carnivori [8-11]. Una delezione di 29 nucleotidi (le unità chimiche che si ripetono e che formano i geni) in ORF 8 era avvenuta in tutti i ceppi isolati durante la media e tarda fase della epidemia [8,9]. La delezione, cioè la perdita, di questi pezzetti del gene portava ad una espressione di due prodotti genici tronchi chiamati ORF8a e ORF8b (Fig. A; in pratica, il gene intero ORF8 si era diviso in 2 parti a seguito della perdita dei 29 pezzetti che rientravano nella sua struttura completa). L’ipotesi maggiormente avanzata inizialmente fu che la formazione di ORF8a ed ORF8b portasse ad una modulazione della patogenicità o della replicazione che favoriva l’adattamento di SARS-CoV all’uomo [8, 10, 11]. Studi piuttosto recenti hanno invece dimostrato che la delezione della ORF8 ha portato ad una attenuazione di SARS-CoV. Il virus, infatti, a seguito della delezione, è diventato meno capace di replicarsi nelle cellule umane [12].
Nelle infezioni naturali, in cui non sono implicate alte dosi del virus, la trasmissione da uomo a uomo generalmente provoca la selezione positiva delle varianti virali meglio adattate all’ospite [13]. Benché la ridotta capacità di replicazione possa in prima analisi apparire come una caratteristica svantaggiosa per il virus, in realtà le varianti nella popolazione virale che hanno minore capacità di replicazione sembrano sopportare meglio ulteriori cambiamenti deleteri per il virus, rispetto alle varianti che mostrano efficiente replicazione nell’ospite [14] e per questo nel tempo vengono selezionate positivamente.Delezioni nei geni accessori sono state osservate anche in MERS-CoV, l’altro coronavirus in grado di infettare l’uomo e provocare sindromi respiratorie severe acute [15-18]. Nonostante non ci sia un dato certo riguardo il ruolo di queste delezioni in Mers-CoV, è da notare che uno dei geni accessori soggetto a delezione, ORF4a, agisce come effettivo antagonista dell’induzione di MDA5 mediata dagli interferoni di tipo I (IFN-I). Gli IFN-I sono proteine prodotte in risposta alle infezioni virali che inducono una vasta gamma di processi tutti volti all’eliminazione del virus. Tra gli enzimi regolati dagli IFN-I vi è appunto MDA5, la cui funzione è degradare i genomi virali; difatti, la replicazione di Mers-CoV in colture di cellule umane dell’epitelio respiratorio si è visto essere fortemente ridotta dal trattamento con IFN-I [19]. Sulla base di questi meccanismi, la delezione di questi geni accessori dovrebbe essere considerata più come forma di attenuazione del virus che come forma di adattamento all’uomo [12].
È anche interessante considerare la conseguenza della propagazione virale conferita da una riduzione della capacità di replicazione del virus. Nel caso del virus Ebola, un prolungato tempo di sopravvivenza del virus (dato da una più lenta replicazione dello stesso) potrebbe incrementare la durata della sua diffusione, portando ad una più durevole permanenza delle varianti virali con un livello di replicazione più basso [20]. Nell’individuo infetto, comunque, una riduzione della replicazione virale si riflette verosimilmente in una attenuazione della patogenicità dell’infezione [12].
COSA STA, INVECE, ACCADENDO A SARS-COV-2 IN TERMINI DI EVOLUZIONE? SI PUÒ RITROVARE LA STESSA DELEZIONE CHE AVREBBE RESO SARS-COV ATTENUATO?
La struttura del genoma di SARS-CoV-2 è piuttosto simile a quella di SARS-CoV (Fig. B), ma presenta alcune differenze nelle proteine accessorie (che abbiamo detto essere quelle più variabili tra i coronavirus). In uno studio riguardante le interazioni tra le proteine di SARS-CoV-2 e quelle umane, viene schematicamente analizzata la omologia tra le proteine di SARS-CoV e SARS-CoV-2 [21]. Come si vede nella Fig. B, in SARS-CoV-2 la regione codificante per la proteina accessoria 8 è intera e nessuna delezione ha portato alla produzione delle due forme tronche 8a ed 8b come invece è stato osservato per SARS-CoV. Tuttavia, l’attuale coronavirus ha subito numerose mutazioni. In particolare, confrontando 7666 sequenze genomiche di SARS-CoV-2, 198 mutazioni sono risultate ricorrenti [4]. La presenza di esse è sinonimo di un adattamento del virus al nuovo ospite, l’uomo. Le mutazioni più ricorrenti in SARS-CoV-2, tuttavia, sembrerebbero non riguardare le proteine accessorie, ma piuttosto quelle non strutturali e la proteina Spike (quella che consente al virus di entrare nelle nostre cellule).
In una recente analisi delle variazioni di sequenza del genoma di SARS-CoV-2, è stata osservata una nuova mutazione, altamente frequente, a carico di un amminoacido della proteina Spike [22]. Ricordiamo che gli amminoacidi sono le unità chimiche di cui sono composte le proteine. La mutazione dell’amminoacido in posizione 614 lungo la sequenza della proteina Spike, definita tecnicamente come D614G, è stata riscontrata nel 70% delle sequenze genomiche di SARS-CoV-2 disponibili a maggio e sembra essere responsabile di un aumento della capacità infettiva del virus. In pratica, la proteina Spike contenente la mutazione D614G renderebbe SARS-CoV-2 più capace di entrare nelle nostre cellule. E questo sembrerebbe deporre male per noi perché, come è ovvio, sorge la domanda: “ALLORA SARS-COV-2 STA DIVENTANDO PIÙ CATTIVO?” Per quanto non banale, la risposta sembra essere NO e questo è molto positivo per noi! Infatti, nonostante SARS-CoV-2 contenente la mutazione D614G sia maggiormente in grado di infettare le nostre cellule rispetto al virus che non presenta questa mutazione, NESSUNA DIFFERENZA NELLA SEVERITÀ DELLA MALATTIA è stata osservata fino ad oggi [23].
PERCHÉ LA MUTAZIONE D614G RENDE SARS-CoV-2 PIÙ INFETTIVO?
Per dare una risposta a questa domanda è necessario spiegare alcuni dei meccanismi biochimici e biomolecolari che il virus utilizza durante la fase di infezione. La proteina Spike usata dal virus per legarsi al recettore ACE 2 presente sulle nostre cellule, si divide in due parti, tecnicamente definiti domini, S1 ed S2.Il dominio S1 è la parte della Spike che interagisce con il recettore ACE2 (in particolare, la regione di S1 che interagisce con ACE2 è quella indicata come RBD nella Fig. C) [24]. Nella regione di giunzione tra il dominio S1 e il dominio S2 (la regione in giallo in Fig. C) è presente un sito polibasico, cioè composto da amminoacidi che hanno carattere basico (nello schema, le lettere “R” in rosso rappresentano un amminoacido chiamato arginina che è un tipico amminoacido basico). Tale sito viene tagliato da un enzima chiamato furina. Il taglio enzimatico è fondamentale per l’attivazione della proteina Spike e per l’ingresso di SARS-CoV-2 nelle nostre cellule [25]. Gli altri coronavirus “SARS-like” isolati dal pipistrello non presentano il sito polibasico e sfruttano altri tipi di enzimi per la separazione dei domini S1 ed S2.
Questa differenza biochimica nel processamento della Spike tra i due virus, SARS-CoV e SARS-CoV-2, è stata indicata come possibile responsabile della maggiore trasmissibilità e diffusione di SARS-CoV-2 rispetto a SARS-CoV [24, 26]. In base agli studi presentati nel lavoro di Zhang L. et al [22], la mutazione D614G limita la dispersione (shedding) del dominio S1 a seguito del taglio enzimatico da parte della furina, stabilizzando l’interazione dello stesso dominio S1 con il dominio S2. L’effetto della mutazione D614G, dunque, rende SARS-CoV-2 maggiormente infettivo riducendo lo shedding di S1 che sarebbe, quindi, maggiormente disponibile per l’aggancio ad ACE-2. Il recettore ACE2 a cui si lega S1, svolge, nelle nostre cellule, più di una fisiologica funzione; la principale è quella di catalizzare la conversione dell’Angiotensina II nel peptide (cioè corta sequenza di amminoacidi) Angiotensina 1-7 [26].
Alcuni studi in corso (dunque non ancora pubblicati, per questo non aggiungiamo referenze) si basano sul fatto che la proteina S1 presenta tutte le caratteristiche di una tossina virale (come accade, ad esempio, per la proteina NS1 del virus Dengue) [27]. Seguendo l’idea che S1 si comporti da tossina virale, è possibile ipotizzare che esista una inibizione del recettore ACE2 mediata da S1. Questa inibizione porterebbe ad uno squilibrio tra l’asse ACE-AngII-ATR1 (pro-trombotico e pro-infiammatorio) e quello ACE2-ANG (1-7)-Mas (anti-infiammatorio e anti-trombotico) [28, 29]. Questa osservazione molto tecnica ci serve per spiegare come la riduzione dello shedding di S1, che potrebbe comportarsi da tossina virale, potrebbe associarsi a una serie di quadri clinici osservati nei pazienti COVID-19. Lasciamo al lettore la comprensione dell’importante significato di questi studi e di quanto la scienza sia vicina a sciogliere ogni nodo relativo a tutti i meccanismi patogenetici di SARS-CoV-2, strada che porta dritta verso un efficiente trattamento della patologia COVID-19.
IN CONCLUSIONE
Sembra dunque che alcune delle mutazioni rilevate in SARS-CoV-2 potrebbero verosimilmente essere soggette a selezione positiva (cioè potrebbero persistere nella popolazione di SARS-CoV-2) e avere effetti sulla interazione di SARS-CoV-2 con l’uomo: per queste ragioni esse andranno monitorate nei successivi sequenziamenti di SARS-CoV-2 e il loro impatto sulla patogenicità del virus andrà attentamente studiato [4, 22].
Ad oggi, dunque, non è possibile trarre conclusioni certe su come SARS-CoV-2 si evolverà in termini di virulenza e quindi se avremo o meno una seconda ondata di COVID-19 così come l’abbiamo vissuta ad inizio pandemia. Tuttavia, visto quanto accaduto per i precedenti due virus SARS-CoV e MERS-CoV, per i quali le mutazioni che sembravano essere solo di adattamento all’ospite, si sono poi rivelate, invece, mutazioni che avevano attenuato il virus circolante nella popolazione umana, ci pare possibile avanzare l’IPOTESI che un destino simile possa esserci per SARS-CoV-2. Una IPOTESI deve poi essere SEMPRE sottoposta a verifica o smentita, e numerosi gruppi di ricerca lavorano ogni giorno al fine di comprendere ogni aspetto riguardante la possibile evoluzione di SARS-CoV-2. Inoltre, bisogna tenere a mente che le interazioni patogeno-ospite tendono naturalmente a evolvere da un lato verso una maggiore infettività e trasmissibilità dell’agente infettivo, dall’altro verso una sua ridotta patogenicità verso l’ospite: una mutazione in grado di aumentare l’infettività di SARS-CoV2 e contemporaneamente ridurne gli effetti patogenetici sarebbe quindi assolutamente in linea con la normale storia naturale dell’adattamento patogeno-ospite. Possiamo quindi avere una CERTEZZA MATEMATICA che l’attuale coronavirus sarà attenuato in una prossima ondata? NO. Si può avanzare l’ipotesi fortemente ragionata che SARS-CoV-2 si presenti in forma meno virulenta in una eventuale seconda ondata? SI! Continuiamo a studiare con realismo ed ottimismo!
Ilaria Baglivo, Biologa, PhD – Università della Campania “Luigi Vanvitelli”
Costanza Maria Cristiani, Biologa, PhD – UNIVERSITÀ “Magna Graecia” di Catanzaro
Walter Lucchesi, Biologo, PhD,- Royal Holloway University of London
Stefano Tasca, MD, Pediatra neonatologo – Casa di Cura città di Roma
Bibliografia
1.Rota, P.A. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome, Science 2003, 300, 1394–1399
2. Drexeler, JF, Corman, VM and Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral research 2014, 101, 45-46
3. Li, KG, Wang W., Zhao XF, Zai JJ, Zhao Q, Li Y et al. Transmission dynamics and evolutionary history of 2019-nCoV J. Med. Virol. 2020, 92, 501-511
4. van Dorp L, Acaman M, Richard D, Shaw LP, Ford CE, Ormond L, Owen CJ, Pang J, Tan CCS, Boshier FAT, Torres Ortis A, Balloux F. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infection, Genetics, Evolution 2020, 83. http://doi.org/10.1016/j.meegid.2020104351
5. Zhang YZ, Holmes EC. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181(2): 223–227.
6. Wu JT et al. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nature Medicine 2020, 26, 506-510
7. Song Z., Xu Y., Bao L., Zhang L, Yu P, Qu Y, Zhu H, X, Zhao W, Han Y and Qin C. From SARS to MERS, Thrusting Coronaviruses Into the Spotlight. Viruses 2019, 11(1):59. doi: 10.3390/v110100599
8. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 2004, 303, 1666–1669.
9. Chiu, R. W. et al. Tracing SARS-coronavirus variant with large genomic deletion. Emerging infectious diseases 2005, 11, 168–170.
10. Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278.
11. Lau, S. K. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 14040–14045.
12.Muth D, Corman VM, Roth H, Binger T, Dijkman R et al Attenuation of replication by 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci Rep 2018, DOI: 10.1038/s41598-018-33487-8
13. Zwart, M. P. & Elena, S. F. Matters of Size: Genetic Bottlenecks in Virus Infection and Their Potential Impact on Evolution. Annu Rev Virol 2015, 2, 161–179
14. Novella, I. S., Elena, S. F., Moya, A., Domingo, E. & Holland, J. J. Size of genetic bottlenecks leading to virus fitness loss is determined by mean initial population fitness. Journal of virology 1995, 69, 2869–2872
15. Payne, D. C. et al. Multihospital Outbreak of a Middle East Respiratory Syndrome Coronavirus Deletion Variant, Jordan: A Molecular, Serologic, and Epidemiologic Investigation. Open Forum Infect Dis 2018, doi: 10.1093/ofid/ofy095
16. Lamers, M. M. et al. Deletion Variants of Middle East Respiratory Syndrome Coronavirus from Humans, Jordan, 2015. Emerging infectious diseases 2016, 22, 716–719.
17. Xie, Q. et al. Two deletion variants of Middle East respiratory syndrome coronavirus found in a patient with characteristic symptoms. Archives of virology 2017, 162, 2445–2449.
18. Lu, X. et al. Spike gene deletion quasispecies in serum of patient with acute MERS-CoV infection. Journal of medical virology 2017, 89, 542–545.
19. Kindler, E. et al. Efficient Replication of the Novel Human Betacoronavirus EMC on Primary Human Epithelium Highlights Its Zoonotic Potential. mBio 4 2013, DOI: 10.1128/mBio.00611-12
20. Marzi, A. et al. Recently Identified Mutations in the Ebola Virus-Makona Genome Do Not Alter Pathogenicity in Animal Models. Cell Rep 2018, 23, 1806–1816.
21. Gordon et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature https://doi.org/10.1038/s41586-020-2286-9.
22. Zhang L et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv, 2020
23. Korber B et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 2020; Zhang L. et al.. bioRxiv 2020
24. J. Shang et al., Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, doi:10.1038/s41586-020-2179-y
25. Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F. Cell entry mechanisms of SARS-CoV-2. PNAS 2020, 117, 11727-11734
26. Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: A Peptidase in the Renin-Angiotensin System, a SARS Receptor, and a Partner for Amino Acid Transporters. Pharmacol Ther. 2010, 128, 119-28. doi: 10.1016/j.pharmthera.2010.06.003.
27. Puerta-Guardo H, Glasner DR, Harris E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12:e1005738. doi: 10.1371/journal.ppat.1005738.
28. Simões e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7)and Mas receptor axis ininflammation and fibrosis. Br J Pharmacol. 2013, 169, 477-92. DOI:10.1111/bph.12159.
29. Rodrigues Prestes TR, Rocha NP, Miranda AS, Teixeira AL, Simoes-E-Silva AC. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence From Basic and Clinical Research. Curr Drug Targets 2017, 18, 1301-1313. doi: 10.2174/1389450117666160727142401.
Leave a Reply
Devi essere connesso per inviare un commento.